[论文解读] PoseBusters: AI-based docking methods fail to generate physically valid poses or generalise to novel sequences
本文提出 PoseBusters,是一个基于 Python RDKit 的测试套件,用于评估蛋白质–配体对接位姿的化学有效性和物理可信度,并显示当前的 DL 基于对接方法在应用这些检查或对新序列泛化时并未优于经典对接工具。
The last few years have seen the development of numerous deep learning-based protein-ligand docking methods. They offer huge promise in terms of speed and accuracy. However, despite claims of state-of-the-art performance in terms of crystallographic root-mean-square deviation (RMSD), upon closer inspection, it has become apparent that they often produce physically implausible molecular structures. It is therefore not sufficient to evaluate these methods solely by RMSD to a native binding mode. It is vital, particularly for deep learning-based methods, that they are also evaluated on steric and energetic criteria. We present PoseBusters, a Python package that performs a series of standard quality checks using the well-established cheminformatics toolkit RDKit. Only methods that both pass these checks and predict native-like binding modes should be classed as having "state-of-the-art" performance. We use PoseBusters to compare five deep learning-based docking methods (DeepDock, DiffDock, EquiBind, TankBind, and Uni-Mol) and two well-established standard docking methods (AutoDock Vina and CCDC Gold) with and without an additional post-prediction energy minimisation step using a molecular mechanics force field. We show that both in terms of physical plausibility and the ability to generalise to examples that are distinct from the training data, no deep learning-based method yet outperforms classical docking tools. In addition, we find that molecular mechanics force fields contain docking-relevant physics missing from deep-learning methods. PoseBusters allows practitioners to assess docking and molecular generation methods and may inspire new inductive biases still required to improve deep learning-based methods, which will help drive the development of more accurate and more realistic predictions.
研究动机与目标
- 推动车辆评估对接预测不仅仅依赖 RMSD,还要结合化学与物理可信度检查的必要性。
- 介绍 PoseBusters,一款基于 RDKit 的测试套件,用于验证配体几何、手性与蛋白–配体相互作用的有效性。
- 在 PoseBusters 标准下比较五种 DL 基础的对接方法和两种经典对接方法。
- 评估对新蛋白靶点的泛化能力以及预测后能量最小化的影响。
提出的方法
- 建立包含三大测试组的 PoseBusters 测试套件:化学有效性与一致性、分子内有效性、分子间有效性。
- 使用 RDKit 进行净化、InChI 标准化、键长/角度检查、平面性测试以及排斥/体积重叠评估。
- 在 Astex Diverse 与 PoseBusters Benchmark 数据集上评估五种 DL 基础的对接方法(DeepDock、DiffDock、EquiBind、TankBind、Uni-Mol)和两种经典方法(AutoDock Vina、Gold)。
- 对已知配体重新对接到同源受体;以 RMSD、PB-有效性与能量检查进行量化。
- 可选择在对接后应用 AMBER ff14sb/Sage/OpenMM 的能量最小化,以评估可行性提升。
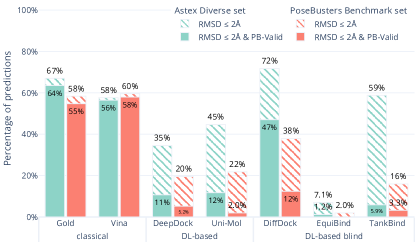
实验结果
研究问题
- RQ1当前的 DL 基础对接方法是否根据 PoseBusters 的检查产生物理上可行的配体位姿?
- RQ2在考虑物理可行性时,DL 基方法与经典对接工具相比表现如何?
- RQ3这些方法对训练阶段未见过的新蛋白靶点的泛化能力如何?
- RQ4对接后能量最小化是否能恢复或改善 DL 基预测的物理可行位姿?
- RQ5在 PoseBusters 标准下,DL 基方法最常见的失效模式是什么?
主要发现
- DL 基于对接方法在 RMSD 取得成功的同时,常常未通过化学/物理可行性检查。
- 在 Astex Diverse 集上,若干 DL 方法的 RMSD≤2 Å,但只有一部分通过所有 PoseBusters 测试(PB-Valid);在考虑可信度时 Gold 与 AutoDock Vina 表现最佳。
- 在 PoseBusters Benchmark 集(新靶点)中,Gold 与 AutoDock Vina 在 RMSD 与 PB-有效性方面再次优于 DL 方法;DiffDock 显示一些 PB-有效位姿,但在 RMSD≤2 Å 的位姿很少。
- DL 方法通常难以对新蛋白进行泛化;当训练数据的靶点序列相似度降低时,其表现下降。
- 对接后能量最小化在 DiffDock、DeepDock、TankBind 与 Uni-Mol 的 PB-有效预测上显著提升,表明 DL 方法缺少力场物理建模;Gold 与 AutoDock Vina 的变化较小。
- PoseBusters 揭示了特定的失效模式(如 Uni-Mol:非标准键长;TankBind:配体内部冲突;EquiBind:蛋白–配体冲突)。
- 总体而言,在同时考虑 RMSD 与物理可行性时,当前没有 DL 基方法胜过经典对接工具。
更好的研究,从现在开始
从论文设计到论文写作,大幅缩短您的研究时间。
无需绑定信用卡
本解读由 AI 生成,并经人工编辑审核。