[论文解读] Towards Predicting Equilibrium Distributions for Molecular Systems with Deep Learning
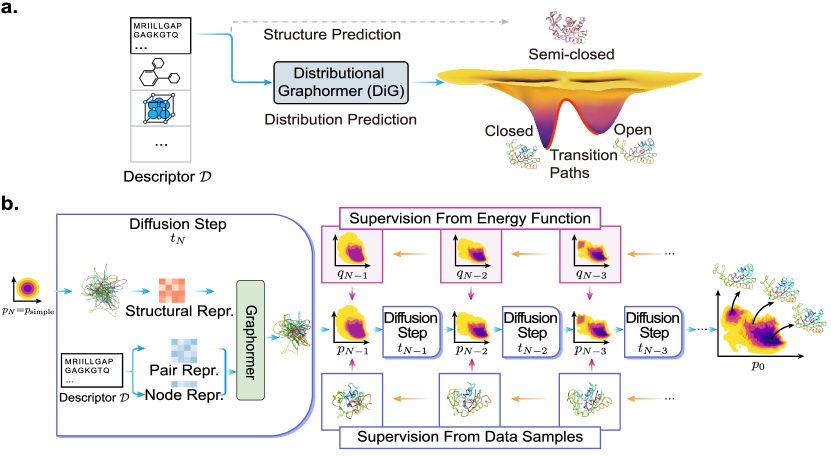
DiG 通过一个受分子描述符条件约束的扩散-based Graphormer 框架,预测分子系统的平衡分布,从而实现对多样构象的高效抽样。
Advances in deep learning have greatly improved structure prediction of molecules. However, many macroscopic observations that are important for real-world applications are not functions of a single molecular structure, but rather determined from the equilibrium distribution of structures. Traditional methods for obtaining these distributions, such as molecular dynamics simulation, are computationally expensive and often intractable. In this paper, we introduce a novel deep learning framework, called Distributional Graphormer (DiG), in an attempt to predict the equilibrium distribution of molecular systems. Inspired by the annealing process in thermodynamics, DiG employs deep neural networks to transform a simple distribution towards the equilibrium distribution, conditioned on a descriptor of a molecular system, such as a chemical graph or a protein sequence. This framework enables efficient generation of diverse conformations and provides estimations of state densities. We demonstrate the performance of DiG on several molecular tasks, including protein conformation sampling, ligand structure sampling, catalyst-adsorbate sampling, and property-guided structure generation. DiG presents a significant advancement in methodology for statistically understanding molecular systems, opening up new research opportunities in molecular science.
研究动机与目标
- 说明需要预测分子系统的平衡分布,而不仅仅是单一结构。
- 提出一个深度学习框架,能够近似平衡分布并抽样多样、化学上可信的结构。
- 实现态密度的估计并支持通过属性引导生成进行逆向设计。
提出的方法
- 引入 Distributional Graphormer (DiG),通过扩散生成将一个简单分布转变为在分子描述符条件下的目标平衡分布。
- 使用前向 Langevin 漂移将目标分布朝向简单高斯分布移动,并基于 Graphormer 学习一个带有分数模型 s^θ_D,t(R) 的反向扩散过程。
- 使用分步、独立监督训练分数模型,可以是基于数据的分数,或利用能量函数 E_D 的物理信息扩散预训练(PIDP)。
- 将扩散过程扎根于分数匹配和扩散理论,通过离散化的反向步获取来自平衡分布的样本 R_0。
- 通过跟踪过程在扩散路径上估计密度,从而计算自由能、熵等热力学量。
- 通过对目标属性 c 条件化来调整分数,从而通过贝叶斯规则的改动转换条件分布 q_D,t(R|c)。
实验结果
研究问题
- RQ1基于扩散的生成模型是否能从描述符近似分子系统的平衡分布?
- RQ2相较于分子动力学模拟和实验结构,DiG 在多样且功能相关的构象抽样方面表现如何?
- RQ3DiG 是否能在配体结合位点取样和催化剂-吸附物吸附分布方面达到与现有方法相当甚至更好的精度?
- RQ4当平衡数据稀缺时,物理信息预训练(PIDP)如何提升学习?
- RQ5DiG 是否能通过对目标属性的条件化来支持属性引导的(逆向)设计?
主要发现
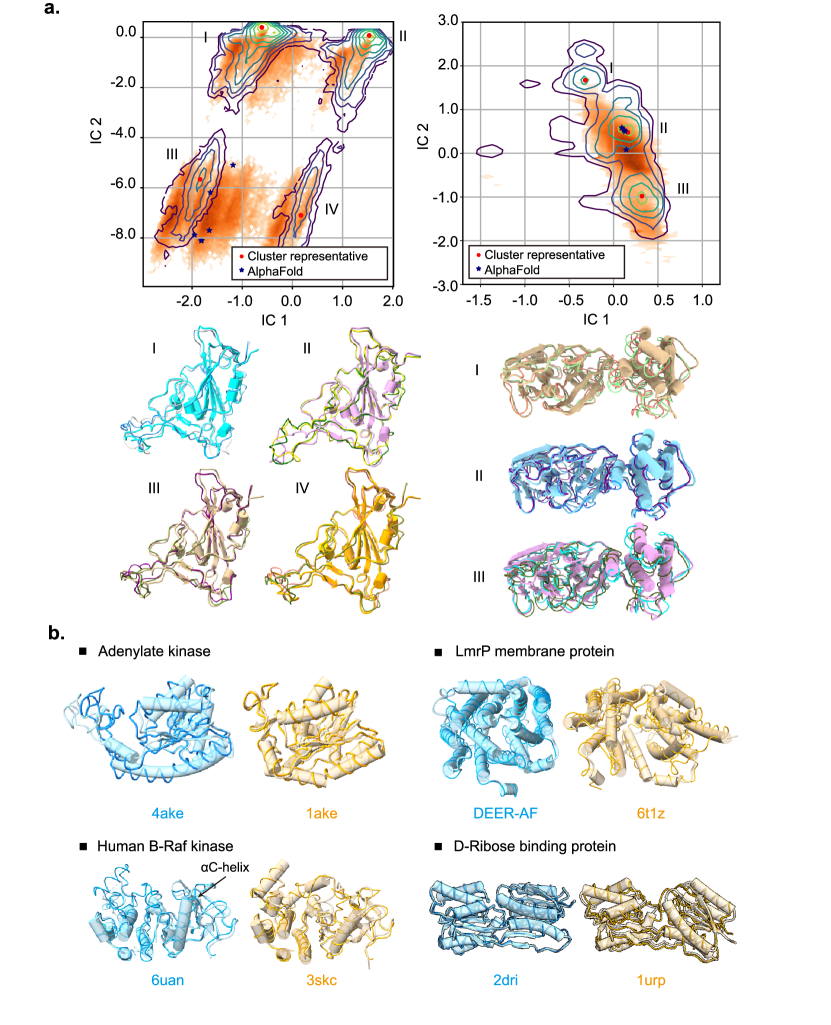
- DiG 可以生成多样的蛋白质构象,类似于在毫秒级 MD 模拟中观察到的分布,并与已知功能状态对齐。
- DiG 在口袋中生成的配体结构的 RMSD 分布表明与晶体结构高度相似,在许多情况下达到约 2.0 Å 的最佳匹配。
- DiG 能在不同吸附位点上抽样催化剂-吸附物吸附构型,紧密匹配基于 DFT 的基线。
- 物理信息扩散预训练(PIDP)通过在预训练阶段利用能量函数来实现分数学习,当平衡数据稀缺时。
- DiG 支持用于属性引导结构生成的条件生成,实现逆向设计,而无需大型条件数据集。
更好的研究,从现在开始
从论文设计到论文写作,大幅缩短您的研究时间。
无需绑定信用卡
本解读由 AI 生成,并经人工编辑审核。