[논문 리뷰] Predicting a Protein's Stability under a Million Mutations
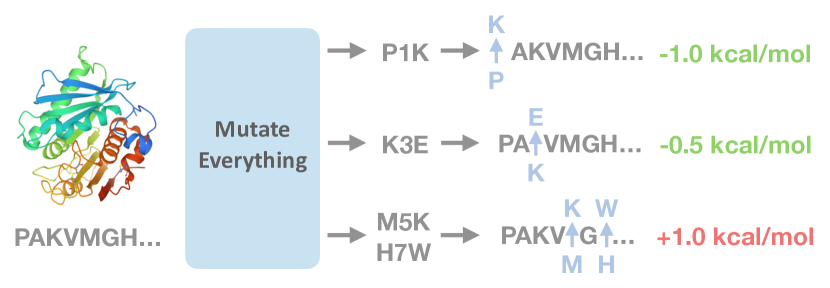
Mutate Everything를 소개하는 병렬 디코딩 방법으로, 단일 및 고차 변이의 ΔΔG를 한 번의 순전파로 예측하여 단백질 전반에 걸친 확장 가능한 안정성 평가를 가능하게 한다.
Stabilizing proteins is a foundational step in protein engineering. However, the evolutionary pressure of all extant proteins makes identifying the scarce number of mutations that will improve thermodynamic stability challenging. Deep learning has recently emerged as a powerful tool for identifying promising mutations. Existing approaches, however, are computationally expensive, as the number of model inferences scales with the number of mutations queried. Our main contribution is a simple, parallel decoding algorithm. Our Mutate Everything is capable of predicting the effect of all single and double mutations in one forward pass. It is even versatile enough to predict higher-order mutations with minimal computational overhead. We build Mutate Everything on top of ESM2 and AlphaFold, neither of which were trained to predict thermodynamic stability. We trained on the Mega-Scale cDNA proteolysis dataset and achieved state-of-the-art performance on single and higher-order mutations on S669, ProTherm, and ProteinGym datasets. Code is available at https://github.com/jozhang97/MutateEverything
연구 동기 및 목표
- 에피스테시스를 고려한 단백질 엔지니어링을 돕기 위한 안정화 변이의 빠른 식별을 촉진한다.
- 한 번의 순전파로 모든 단일 및 고차 변이에 대해 ΔΔG를 계산하는 확장 가능한 디코더를 개발한다.
- 벤치마크 안정성 데이터셋(ProTherm, S669, ProteinGym)에서 최첨단 성능을 입증한다.
- 단백질 전반에 걸쳐 변이를 열거할 수 있는 실용적 확장성을 보이고 기존 방법과 비교해 유리한 성능을 보여준다.
제안 방법
- 사전 학습된 백본(AlphaFold, 미세조정; ESM2도 사용할 수 있음)을 사용하여 서열의 위치별 특징을 추출한다.
- 각 위치와 가능한 아미노산에 대해 잠재 변이 표현 z(μ)=f^t(x_p)+h^t를 계산한다.
- 가벼운 머리 g^1으로 단일 변이를 디코딩하여 z(μ)로부터 ΔΔG를 예측한다.
- 고차 변이 M={μ_k}에 대해 단일 변이 표현들을 합산(합)하고 가벼운 머리 g를 사용해 ΔΔG를 예측하도록 디코딩한다.
- L×20 가능한 모든 변이에 대해 z(μ)를 미리 계산해 두고, 어떤 변이 집합에 대해서도 해당 z(μ)를 인덱싱해 합산한 뒤 병렬로 디코딩한다.
- 단일 및 이중 변이에 대해 허버 손실(Huber loss)로 학습하고 필요에 따라 고차 상호작용의 잔차를 학습한다.
실험 결과
연구 질문
- RQ1하나의 순전파로 단일 및 고차 변이에 대한 ΔΔG를 정확하게 예측하는 통합형 병렬 디코더가 가능한가?
- RQ2Mutate Everything 프레임워크가 ProTherm, S669, ProteinGym 등 기존의 안정성 벤치마크에서 최첨단 방법과 비교해 어떤 성능을 보이는가?
- RQ3한 번의 순전파에서 모든 변이를 열거하는 것이 큰 단백질이나 전체 단백질체에 대한 안정성 분석을 실현 가능한가?
- RQ4 AlphaFold/ESM2와 같은 백본의 미세조정이 구조 좌표 없이 안정성 예측을 얼마나 개선하는가?
- RQ5고차 변이 집합에서의 에피스타시스(상호작용 효과)를 모델이 얼마나 잘 다루는가?
주요 결과
| 방법 | r_s | AUC | MCC | RMSE ↓ | 안정화 r_s |
|---|---|---|---|---|---|
| Mutate Everything (Ours) | 0.53 (0.01) | 0.78 (0.01) | 0.43 (0.01) | 2.04 (0.01) | 0.19 (0.01) |
| Mutate Everything (Additive) | 0.50 (0.02) | 0.76 (0.01) | 0.37 (0.02) | 2.02 (0.03) | 0.20 (0.01) |
| PROSTATA (Additive) | 0.50 (0.00) | 0.73 (0.02) | 0.28 (0.02) | 1.44 (0.02) | 0.00 (0.00) |
- 프로Therm PTMul에서의 Spearman 상관계수 0.53, S669에서 0.56으로 최첨단 성능을 달성했고 ProteinGym 안정성 벤치마크에서 0.52–0.53의 r_s 및 AUC/MCC 지표에서 우수한 성능을 보인다.
- 이중 변이의 안정화를 비안정화 변이보다 앞서 순위를 매겨 nDCG 0.43 대 0.25, DetPr 0.16 대 0.10를 기록했다(cDNA2에서).
- ESM2 백본으로는 0.6초, AlphaFold 백본으로는 12.1초 수준으로 한 GPU에서 모든 단일 및 이중 변이를 한 번의 순전파로 실행한다.
- 단순 합산이 아닌 에피스타시스를 모델링함으로써/additive 베이스라인 대비 향상된 성능을 보인다.
- ProteinGym 및 ProteinGym-Stability로의 일반화가 가능하며, 앙상블링으로 Tranception 기반 방법 대비 이득을 얻는다.

더 나은 연구,지금 바로 시작하세요
연구 설계부터 논문 작성까지, 연구 시간을 획기적으로 줄여보세요.
카드 등록 없음 · 무료 플랜 제공
이 리뷰는 AI가 만들고, 인간 에디터가 검토했습니다.