[논문 리뷰] The ab initio amorphous materials database: Empowering machine learning to decode diffusivity
본 논문은 AIMD를 사용하여 가장 큰 계산된 비정질 재료 데이터베이스(5120 조성, 79 원소)를 구축하고, Li+ 확산도 예측을 위한 ML 모델(RF, XGBoost, SISSO)을 시연하며, M3GNet을 통한 대리구조 생성으로 보완한다.
Amorphous materials exhibit unique properties that make them suitable for various applications in science and technology, ranging from optical and electronic devices and solid-state batteries to protective coatings. However, data-driven exploration and design of amorphous materials is hampered by the absence of a comprehensive database covering a broad chemical space. In this work, we present the largest computed amorphous materials database to date, generated from systematic and accurate extit{ab initio} molecular dynamics (AIMD) calculations. We also show how the database can be used in simple machine-learning models to connect properties to composition and structure, here specifically targeting ionic conductivity. These models predict the Li-ion diffusivity with speed and accuracy, offering a cost-effective alternative to expensive density functional theory (DFT) calculations. Furthermore, the process of computational quenching amorphous materials provides a unique sampling of out-of-equilibrium structures, energies, and force landscape, and we anticipate that the corresponding trajectories will inform future work in universal machine learning potentials, impacting design beyond that of non-crystalline materials.
연구 동기 및 목표
- 이온 전도체의 데이터 기반 발견을 가속하기 위한 포괄적 비정질 재료 데이터베이스의 필요성을 제고한다.
- 폭넓은 조성 및 온도에 걸친 큰 규모의 다양하고 AIMD 유래의 비정질 데이터베이스를 생성한다.
- 조성 및 구조 관련 특징으로 학습된 머신러닝 모델을 활용해 Li 이온 확산도를 빠르게 예측할 수 있도록 한다.
제안 방법
- MPMorph 워크플로를 사용해 5,120개 조성의 비정질 구조를 5000 K에서 융해시켜 생성한다.
- 선정된 조성에 대해 마지막 스냅샷 구조를 1000–2500 K에서 어닐링하여 두 번째 다온도 데이터베이스를 만든다.
- AIMD 기반 궤적에서 Li 확산도와 활성화 에너지를 계산하고 ML용 풍부한 특징 집합을 추출한다.
- 온도 의존 확산도를 예측하기 위해 Random Forest와 XGBoost 모델을 학습시키고 다섯 겹 교차검증으로 일반화를 평가한다.
- Li 확산도를 예측하기 위한 SISSO 디스크립터를 개발하고 모델 복잡도에 따른 성능을 비교한다.
- ML 입력을 위한 구조 특징을 계산하고 대리구조 생성을 위한 대리모델로 M3GNet를 통합하며 속도 향상과 적용 가능성을 평가한다.

실험 결과
연구 질문
- RQ1크고 다양한 계통의 ab initio 비정질 재료 데이터베이스가 조성과 온도에 걸쳐 Li+ 확산도의 정확한 ML 예측을 가능하게 할 수 있는가?
- RQ2비정질 시스템에서 Li 확산도와 활성화 에너지와 가장 강하게 상관하는 조성 및 구조 특징은 무엇인가?
- RQ3앙상블 모델(RF, XGBoost)과 SISSO 디스크립터가 비정질 데이터로부터 Li 확산도를 예측하는 데 어떤 차이를 보이는가?
- RQ4보편적 ML 포텐셜(M3GNet/CHGNet)이 비정질 구조를 신뢰성 있게 생성하고 확산도 예측에 유용한 입력을 제공할 수 있는가?
주요 결과
- 데이터베이스는 5120 개의 조성과 79 개 원소를 포괄하며, 5000 K 비정질 세트에서 Li 함유 화합물이 3,533개이다.
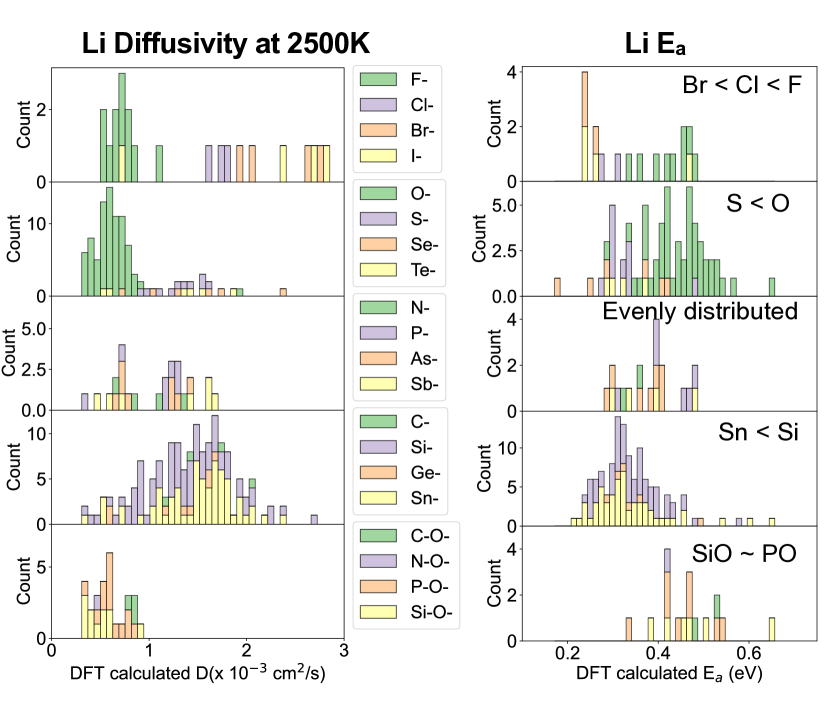
- Li 확산도와 활성화 에너지 분포는 음이온 유형, 양이온 크기 및 전기음성도 차이에 따라 명확한 경향을 보인다.
- RF와 XGBoost는 확산도 예측에서 R^2가 1에 가까운 거의 완벽한 적합을 달성하고 낮은 MAE와 RMSE를 보이며 교차검증으로 검증됐다.
- 최고 예측 특징에는 Li%와 Li 외 환경 및 결합을 설명하는 특징들, 예를 들면 평균 응집에너지, 포장 분율, Li 이웃 개수 등이 포함된다.
- SISSO 모델은 최적의 3-디스크립터로 확산도를 예측할 수 있으며, 온도, 조성 및 구조 간 해석 가능한 관계를 강조한다.
- M3GNet은 AIMD 유도 구조를 높은 충실도로 재현할 수 있으며 RDF에 대해 R^2 최대 0.99에 이르고 구조 생성을 약 2000배 가속시키나 고온(5000 K) 확산도에는 덜 정확하다.
더 나은 연구,지금 바로 시작하세요
연구 설계부터 논문 작성까지, 연구 시간을 획기적으로 줄여보세요.
카드 등록 없음 · 무료 플랜 제공
이 리뷰는 AI가 만들고, 인간 에디터가 검토했습니다.