[論文レビュー] Denoising Diffusion Probabilistic Models
この論文は拡散確率モデルを用いた高品質画像合成を紹介し、それらをデノイジングスコアマッチングと Langevin ダイナミクスと結びつけ、CIFAR-10 でのFIDの最先端と CelebA-HQ および LSUN での高品質サンプルを示す。
DiffuCpG 1. Introduction In this study, we used a generative AI diffusion model to address missing methylation data. We trained the model with Whole-Genome Bisulfite Sequencing data from 26 acute myeloid leukemia samples and validated it with Reduced Representation Bisulfite Sequencing data from 93 myelodysplastic syndrome and 13 normal samples. Additional testing included data from the Illumina 450k methylation array and Single-Cell Reduced Representation Bisulfite Sequencing on HepG2 cells. Our model, DiffuCpG, outperformed previous methods by integrating a broader range of genomic features, utilizing both short- and long-range interactions without increasing input complexity. It demonstrated superior accuracy, scalability, and versatility across various tissues, diseases, and technologies, providing predictions in both binary and continuous methylation states. In this repository, we deposit the code used to build the diffusion models along with necessary example datasets to train and test a diffusion model for methylation imputation purposes. Docker Usage Install Docker Install Docker using the following link:https://docs.docker.com/engine/install/Recommended system specs: Debian 12 bookworm with 16GB RAM or more.Make sure you have the latest Nvidia GPU driver installed and docker can access your Nvidia GPU. Run Docker images with Tissue-specific Models docker pull yay135/diffucpg_tssUse our example to generate input samples with Hi-C matrix and CIS (Confidence Interval Cross Sample) data.docker run -it yay135/diffucpg_tssthenpython generate_train_test_samples.py The tissue-specific models (pytorch) are for CD34+ cells, GBM and BRCA, they are stored in folders named "model*" in the image. Run the Tissue specific modelsdocker run -it yay135/diffucpg_tssthenpython batch_run.py Run Docker images Example Models docker pull yay135/diffucpgIf you do not have a GPU enabled system, pull a CPU-only imagedocker pull yay135/diffucpg_cpuprepare your input data directory, use the following command to print a example input data directorydocker run --rm yay135/diffucpg -e trueassume your data directory name is "input_data"in windowsdocker run --gpus all -v .\input_data\:/data --rm yay135/diffucpgin unix or linuxdocker run --gpus all -v ./input_data:/data --rm yay135/diffucpg Other docker options -d or --device : select which cuda device to run with, default is 0-m or --mingcpg : scan your methyl array, limit only imputing windows with at least m non-missing methyl values, default is m=10-o or --overlap : set number of impute epochs, shift window locations between epochs, get mean imputed values for each CpG location, default is 2example:docker run --gpus all -v ./input_data:/data --rm yay135/diffucpg -d 1 -m 5 -o 3use cuda device 1, min number of non-missing methyl values in a window is 5, overlap epochs 3 The following tutorials are for non-docker usages. 2. Data and Models Example datasets are available for download using "gdown.sh". The example datasets only contain WGBS methylation data. The model is the DDPM diffusion model, the repository contains a complete implementation for 1-dimensional input. Please refer to https://arxiv.org/abs/2006.11239 and https://huggingface.co/blog/annotated-diffusion for more details. 3. How to use 3.1 System Requirements The number of steps in the diffusion process is set to 2000. Imputing a sample requires 2000 steps. Gpu acceleration is preferred. 16GB of RAM is required. The code is fully tested and operational on the following platform: Distributor ID: DebianDescription: Debian GNU/Linux 12 (bookworm)Release: 12Codename: bookworm 3.2 Clone the Current Project Run the following command to clone the project.git clone https://github.com/yay135/DiffuCpG.git 3.4 Configure Environment Make sure you have the following software installed in your system:Python 3.9+Pytorch 2.0.1+ 3.4 Run Training and Testing python run.pyThe script will download necessary data and install dependencies automatically. 4 Data and Script Details 4.1 RAW Data The methylation arrays downloaded are in the folder "raw", each file is a methylation array. The first 2 columns are "chromosome" and "location". The assembly used for mapping in our project is the "GRCH37 primary assembly". It is also downloaded automatically. The rest of the columns in each file are methylation levels(required) and other biological data (optional) you wish to incorporate to enhance the model. These files in the raw folder are the initial inputs for pipeline,if you wish to use your own data, it must be configured as such before running the pipeline. 4.2 Generate Sample Use script "generate_samples.py" to generate samples for training and testing.The model can not directly read and impute a methylation array file. Instead, each methylation array is divided into windows, each window is 1kb (1000 base pairs) in length, and each training testing sample is generated from a window. Each sample contains at least 5 channels. the first 4 is the sequence one-hot encoding, the 5th is the methylation data. If a base pair location is not a CpG location, the methylation data value for it is "-1". If a CpG's methylation data is missing or waiting for imputaion, its value is also "-1". Other biological data can be added as extra channels. Check out example raw files in the folder "raw" to form your own datasets for training and testing sample generation.For each raw file in the "raw" folder, the first 3 columns are chr, loc, and methylation.The rest of the columns are treated as additional channels and will be added to each sample during generation. '-d' or '--folder': specify raw data folder'-i' or '--index' : which column in a raw file is the methylation array'-t' or '--tol' : how many missing methylation value is tolerated(we recommend 0 for generating training samples and -1 for generating testing samples, 0 will force the script to only select from windows with no missings, -1 will tolerate missing as much as possible.)'-c' or '--chr' : limit which chromosome to use, default is "chr#" to use all chromosomes'-w' or '--winsize' : what window size to use, default is 1000 '-m' or '--mincpg': force generate from window to have a minimum number of CpGs, default is 10 '-n' or '--nsample': number of samples to generate per chromosome '-p' or '--output': samples output folder, default is "out" Use script "generate_samples_concat.py" to generate samples from long-range interacting windows such as Hi-C interactions or computed correlation.Check out the example long range file in the folder "data" to form your own long-range interacting windows for sample generation and concatenation. 4.3 Training Script Use diffusion.py to train and test a DDPM model using the generated samples'-t' or '--train_folder' : the folder containing the training samples'-f' or '--model_folder' : the model folder, will be created if it does not exist'-w' or '--win_size' : window size of each sample, default is 1000'-c' or '--channel': channel size of each sample'-d' or '--cuda_device' : if you have multiple cuda gpus, select which gpu to use, default is 0"-e" or "--epoch" : how many epochs for training, default is 2000"-s" or "--earlystop" : whether to use "early stopping" during training, default is False"-p" or "--patience" : patience for early stopping, default is 10 4.4 Imputation Use diffusion_inpainting.py to perform imputation on generated samples.'-t' or '--test_folder' : the folder containing samples for imputation'-o' or '--out_folder': imputed output folder name, default="inpainting_out"'-w' or '--win_size' : window size of each sample, default is 1000'-c' or '--channel': channel size of each sample'-d' or '--cuda_device' : if you have multiple cuda gpus, select which gpu to use, default is 0 Team If you have any questions or concerns about the project, please contact the following team member: Fengyao Yan fxy134@miami.edu
研究の動機と目的
- 拡散モデルを画像合成のための単純で訓練可能な潜在変数モデルのクラスとして動機づける。
- デノイジングスコアマッチングの視点が現実的な訓練目的とサンプリング手順をもたらすことを示す。
- CIFAR-10、CelebA-HQ、および LSUN データセットで高品質なサンプル生成を実証する。
- 拡散モデルのレート歪み特性と段階的デコード能力を分析する。
提案手法
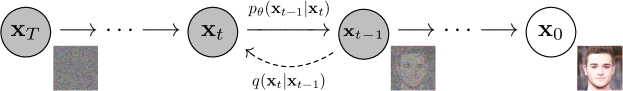
- 固定された順向マルコフ連鎖を持つ拡散モデルを定義し、徐々にガウスノイズを加える。
- 逆過程をガウス条件付きでパラメータ化し、Sigmaを固定定数として選択する。muのパラメータ化を探る。
- epsilon-predictor逆パラメータ化とアニーリング Langevin ダイナミクスを用いたデノイジングスコアマッチングとの同値性を示す。
- ノイズレベルを横断するデノイジングスコアマッチングと整合する簡略化された訓練目的 L_simple を導入する。
- 最終デコーダを離散化して離散データ対数尤度を可能にし、CIFAR-10、CelebA-HQ、LSUN で評価する。

実験結果
リサーチクエスチョン
- RQ1拡散確率モデルは標準ベンチマークで既存の生成モデルと同等以上の高品質な画像を生成できるか?
- RQ2逆過程の異なるパラメータ化と訓練目的がサンプル品質と尤度に与える実践的影響は何か?
- RQ3拡散フレームワークはデノイジングスコアマッチングと Langevin ダイナミクスとどのように関連し、この結びつきからどんな利点が生まれるか?
- RQ4ロス圧縮に対する拡散モデルのレート歪み特性と段階的デコード能力はどのようなものか?
- RQ5拡散モデルは逐次サンプリングとデコードを通じて自己回帰型のデコード利点をサポートするか?
主な発見
| モデル | IS | FID | NLL テスト(訓練) |
|---|---|---|---|
| Ours (L_simple) | 9.46±0.11 | 3.17 | ≤3.75 (3.72) |
| Ours (L, fixed isotropic Σ) | 7.67±0.13 | 13.51 | ≤3.70 (3.69) |
- CIFAR-10 の無条件生成で、簡略化された訓練目的で Inception Score 9.46 および FID 3.17 を達成。
- 最良モデルを用いた無条件の CIFAR-10 結果は、クラス条件付きモデルと比較してもサンプル品質で多くの既存手法を凌駕する。
- epsilon-prediction 逆パラメータ化は変分下界で訓練した場合 mu-prediction に匹敵し、簡略化された目的でははるかに優れている。
- LSUN 256×256 および CelebA-HQ の高視覚品質サンプルで競争力のあるまたは優れた FID スコアをもたらす。
- モデルのレート歪み分析は、ほとんどの lossless ビットが知覚できない画像細部を特徴とすることを明らかにし、強力な段階的ロスィーコーディング挙動を示唆する。
- 段階的デコード実験は、大規模な画像特徴がサンプリング過程で早期に現れ、細部は後で現れることを示している。
より良い研究を、今すぐ始めましょう
論文設計から論文執筆まで、研究時間を劇的に削減しましょう。
クレジットカード登録不要
このレビューはAIが作成し、人間の編集者が確認しました。