[論文レビュー] EspalomaCharge: Machine learning-enabled ultra-fast partial charge assignment
EspalomaChargeは、ハイブリッドなグラフ神経ネットワークと電荷均衡化を用いて、AM1-BCC ELF10様の部分電荷をO(N)スケーリングで予測し、小分子および生体高分子のコンフォマーに依存しない高速な充電を実現します。参照精度に匹敵し、一般的なワークフローへのドロップイン置換として統合されます。
Atomic partial charges are crucial parameters in molecular dynamics (MD) simulation, dictating the electrostatic contributions to intermolecular energies, and thereby the potential energy landscape. Traditionally, the assignment of partial charges has relied on surrogates of \textit{ab initio} semiempirical quantum chemical methods such as AM1-BCC, and is expensive for large systems or large numbers of molecules. We propose a hybrid physical / graph neural network-based approximation to the widely popular AM1-BCC charge model that is orders of magnitude faster while maintaining accuracy comparable to differences in AM1-BCC implementations. Our hybrid approach couples a graph neural network to a streamlined charge equilibration approach in order to predict molecule-specific atomic electronegativity and hardness parameters, followed by analytical determination of optimal charge-equilibrated parameters that preserves total molecular charge. This hybrid approach scales linearly with the number of atoms, enabling, for the first time, the use of fully consistent charge models for small molecules and biopolymers for the construction of next-generation self-consistent biomolecular force fields. Implemented in the free and open source package \texttt{espaloma\_charge}, this approach provides drop-in replacements for both AmberTools \texttt{antechamber} and the Open Force Field Toolkit charging workflows, in addition to stand-alone charge generation interfaces. Source code is available at \url{https://github.com/choderalab/espaloma_charge}.
研究の動機と目的
- 大規模な生体分子にスケーラブルで、コンフォマーに依存せず高速かつ高精度な部分電荷割り当て法を開発する。
- グラフニューラルネットワークを活用して、電荷平衡化のための原子レベルの電気陰性度と硬さパラメータを予測する。
- 予測された電荷の総和が分子全体の総電荷Qになるよう、解析的な制約付き解によって保証する。
- 既存の分子力学ワークフロー(AmberTools、Open Force Field Toolkit)への容易な統合を提供する。
- この方法がQMベースのアプローチに比べて低コストでAM1-BCC ELF10品質の電荷を達成することを実証する。
提案手法
- 原子環境の連続埋め込みを生成するグラフニューラルネットワークを構築する(Espalomaフレームワーク)。
- GNN埋め込みから各原子の制約なしの電気陰性度 e_i および硬さ s_i を予測する。
- 和を最小化して各原子の電荷 q_i を解析的に解く。目的関数は sum_i (e_i q_i + 0.5 s_i q_i^2)、制約条件は sum_i q_i = Q(総分子電荷)。
- 拡張SPICEデータセットで訓練し、二乗誤差損失でAM1-BCC ELF10電荷を再現する。
- O(N) 実行時間計算量と大規模分子セットをバッチ処理できる能力を示す。
- OpenFF ToolkitとAmberワークフローへの統合のためのPython APIとCLIを提供する。

実験結果
リサーチクエスチョン
- RQ1多様な化学空間にわたって、機械学習の代理モデルがAM1-BCC ELF10電荷をどれだけ正確に再現できるか?
- RQ2大規模系に対して、EspalomaChargeの計算スケーリングと速度はAmberToolsおよびOpenEyeと比較してどうか?
- RQ3EspalomaChargeはトレーニング分布の外にある生体分子および薬物様化合物に対して一般化するか?
- RQ4EspalomaChargeは既存のMM/MDワークフロー内でドロップイン方式で電荷を提供できるか?
主な発見
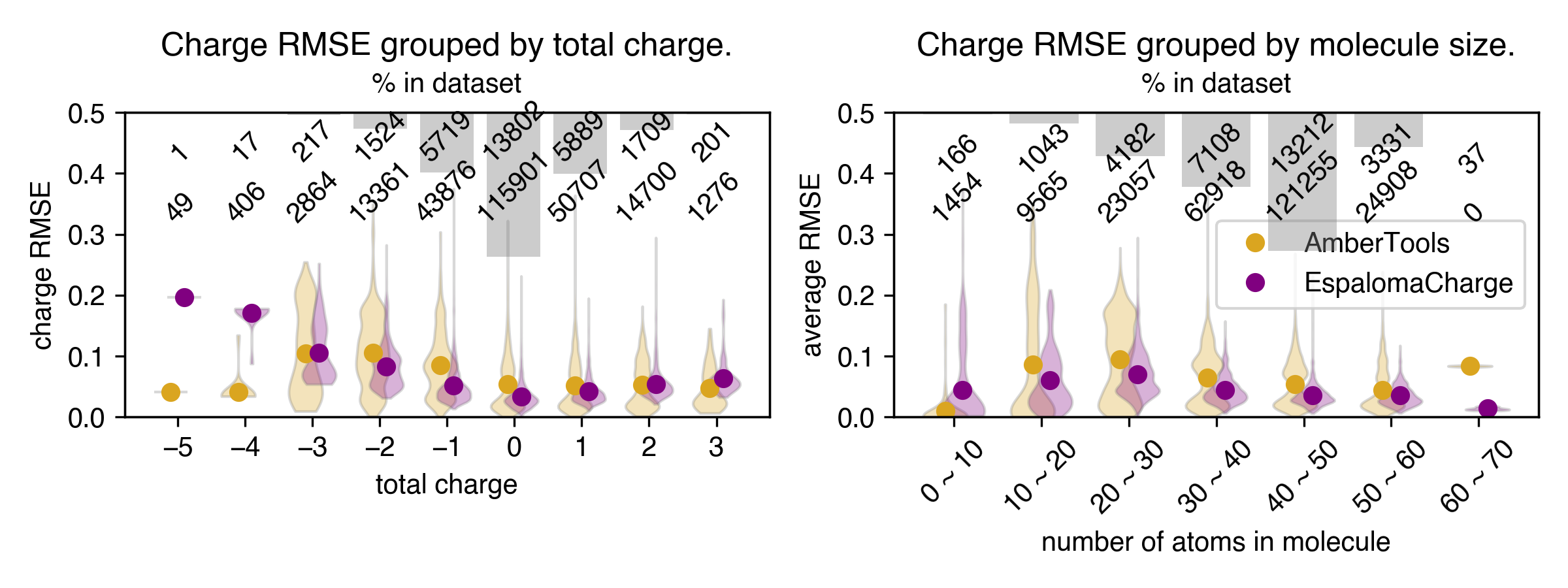
- EspalomaChargeは AM1-BCC ELF10電荷を、AM1-BCC実装間の差と同程度のRMSEで再現する(しばしばAmberToolsとOpenEyeの比較と同等)。
- SPICEテストセットでは、RMSE ≈ 0.0435 and walltime ≈ 93.10 s (EspalomaCharge)、他のベースラインより高速、OpenEyeとAmberToolsはさまざまな文脈でより長い時間を示す。
- 複数のデータセット(FDA-approved, ZINC250K, FreeSolv, PDB eXpo)で、EspalomaChargeはRMSE値を0.0110–0.0266の範囲で達成し、化学空間全体で堅牢な精度を示す。
- EspalomaChargeは原子数に対して線形時間で動作(O(N))、QMベースの充電法より桁違いに高速で、生体高分子(数百残基)のパラメータ化を数秒で可能にする。
- 単一の電荷計算で多くの分子をバッチ処理すると、CPU/GPUで大幅な速度向上を得られ、実用的なライブラリサイズでほぼ一定時間に近づく。
- EspalomaCharge電荷を用いた水和自由エネルギー計算は、RMSEと決定係数R^2の点で実験と比較してAmberToolsおよびOpenEyeの実装と統計的に区別不能である。
より良い研究を、今すぐ始めましょう
論文設計から論文執筆まで、研究時間を劇的に削減しましょう。
クレジットカード登録不要
このレビューはAIが作成し、人間の編集者が確認しました。