[論文レビュー] Predicting a Protein's Stability under a Million Mutations
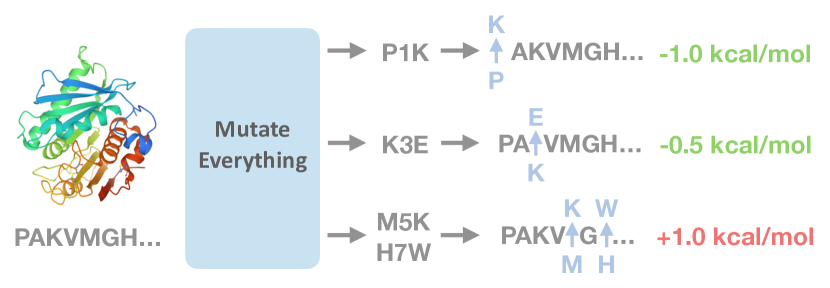
Mutate Everything を導入する。これは全ての単一および高次の変異を1回の前方伝播でΔΔGを予測する並列デコーディング手法であり、タンパク質全体にわたるスケーラブルな安定性評価を可能にする。
Stabilizing proteins is a foundational step in protein engineering. However, the evolutionary pressure of all extant proteins makes identifying the scarce number of mutations that will improve thermodynamic stability challenging. Deep learning has recently emerged as a powerful tool for identifying promising mutations. Existing approaches, however, are computationally expensive, as the number of model inferences scales with the number of mutations queried. Our main contribution is a simple, parallel decoding algorithm. Our Mutate Everything is capable of predicting the effect of all single and double mutations in one forward pass. It is even versatile enough to predict higher-order mutations with minimal computational overhead. We build Mutate Everything on top of ESM2 and AlphaFold, neither of which were trained to predict thermodynamic stability. We trained on the Mega-Scale cDNA proteolysis dataset and achieved state-of-the-art performance on single and higher-order mutations on S669, ProTherm, and ProteinGym datasets. Code is available at https://github.com/jozhang97/MutateEverything
研究の動機と目的
- エピスタシスの下でのタンパク質工学を支援する安定化変異の迅速な同定を促進する。
- 1回の前方伝播で全ての単一および高次の変異についてΔΔGを計算するスケーラブルなデコーダを開発する。
- ベンチマーク安定性データセット(ProTherm、S669、ProteinGym)で最先端の性能を示す。
- タンパク質全体にわたって変異を列挙する実用的なスケーラビリティを示し、既存手法と比較して有利であることを示す。
提案手法
- 事前学習済みバックボーン(Fine-tuned の AlphaFold; あるいは ESM2 も使用可能)を用いて配列の各位置の特徴を抽出する。
- 各位置と可能なアミノ酸に対して潜在変異表現 z(μ)=f^t(x_p)+h^t を計算する。
- 軽量ヘッド g^1 を用いて z(μ) から ΔΔG を予測する。
- 高次の変異 M={μ_k} について、単一変異表現を集約(和)し、軽量ヘッド g で ΔΔG を予測する。
- L×20 の全ての変異 z(μ) を事前計算する。任意の変異集合について、該当する z(μ) を索引付けして和を取り、並列にデコードする。
- 単一および二重変異体を対象にHuber損失で学習し、必要に応じて高次相互作用の残差を学習する。
実験結果
リサーチクエスチョン
- RQ1統一された並列デコーダは、単一および高次の変異の双方について正確に ΔΔG を予測できるか。
- RQ2Mutate Everything フレームワークは、確立された安定性ベンチマーク(ProTherm、S669、ProteinGym)で最先端手法と比較してどのような性能を示すか。
- RQ3すべての変異を1回の前方伝播で列挙することは、巨大なタンパク質や全プロテオームにわたる安定性解析を実現可能にするか。
- RQ4構造座標なしで、AlphaFold/ESM2 のようなバックボーンをファインチューンすることで安定性予測をどの程度改善できるか。
- RQ5高次変異セットにおけるエピスタシス効果をモデルはどのように処理するか。
主な発見
| Method | r_s | AUC | MCC | RMSE ↓ | Stabilizing r_s |
|---|---|---|---|---|---|
| Mutate Everything (Ours) | 0.53 (0.01) | 0.78 (0.01) | 0.43 (0.01) | 2.04 (0.01) | 0.19 (0.01) |
| Mutate Everything (Additive) | 0.50 (0.02) | 0.76 (0.01) | 0.37 (0.02) | 2.02 (0.03) | 0.20 (0.01) |
| PROSTATA (Additive) | 0.50 (0.00) | 0.73 (0.02) | 0.28 (0.02) | 1.44 (0.02) | 0.00 (0.00) |
- ProTherm PTMul (0.53) および S669 (0.56) で最先端の Spearman 相関を達成し、ProteinGym 安定性ベンチマーク(0.52–0.53 rs)および AUC/MCC 指標で上回る。
- cDNA2 で、安定化二重変異を不安定化より前にランク付けし、nDCG 0.43 対 0.25、DetPr 0.16 対 0.10。
- 単一GPUで、すべての単一および二重変異を1回の前方伝播で数秒で実行(ESM2 で 0.6s、AlphaFold バックボーンで 12.1s)。
- 加法的ベースラインを上回るのは、単純な単一変異の和ではなくエピスタシスをモデル化するため。
- ProteinGym および ProteinGym-Stability への汎化を示し、アンサンブルにより Tranception ベースの手法より利得を得る。

より良い研究を、今すぐ始めましょう
論文設計から論文執筆まで、研究時間を劇的に削減しましょう。
クレジットカード登録不要
このレビューはAIが作成し、人間の編集者が確認しました。