[論文レビュー] The ab initio amorphous materials database: Empowering machine learning to decode diffusivity
本研究はAIMDを用いて、計算による最大規模の非晶質材料データベース(5120組成、79元素)を構築し、Li+拡散度を予測する機械学習モデル(RF、XGBoost、SISSO)を示し、M3GNetによる代理構造生成を併用する。
Amorphous materials exhibit unique properties that make them suitable for various applications in science and technology, ranging from optical and electronic devices and solid-state batteries to protective coatings. However, data-driven exploration and design of amorphous materials is hampered by the absence of a comprehensive database covering a broad chemical space. In this work, we present the largest computed amorphous materials database to date, generated from systematic and accurate \textit{ab initio} molecular dynamics (AIMD) calculations. We also show how the database can be used in simple machine-learning models to connect properties to composition and structure, here specifically targeting ionic conductivity. These models predict the Li-ion diffusivity with speed and accuracy, offering a cost-effective alternative to expensive density functional theory (DFT) calculations. Furthermore, the process of computational quenching amorphous materials provides a unique sampling of out-of-equilibrium structures, energies, and force landscape, and we anticipate that the corresponding trajectories will inform future work in universal machine learning potentials, impacting design beyond that of non-crystalline materials.
研究の動機と目的
- データ駆動型のイオン伝導体の発見を加速するために、包括的な非晶質材料データベースの必要性を動機づける。
- 幅広い組成と温度をカバーする、largeで多様なAIMD由来の非晶質データベースを生成する。
- 組成・構造に関連する特徴を用いて訓練された機械学習モデルを用い、Liイオン拡散度の迅速な予測を可能にする。
提案手法
- MPMorphワークフローを用いて、5,120組成の非晶構造を5000 Kで融解させて生成する。
- 選択された組成について、最後のスナップショット構造を1000–2500 Kでアニーリングして、別の多温度データベースを作成する。
- AIMDベースの軌道からLi拡散係数と活性エネルギーを計算し、機械学習のための豊富な特徴セットを抽出する。
- Random ForestとXGBoostモデルを訓練して温度依存性拡散度を予測し、五分割交差検証による汎化性能を評価する。
- SISSO記述子を開発し、Li拡散度を予測し、モデルの複雑さ間で性能を比較する。
- 構造の代理としてM3GNetを統合し、非晶構造を生成してML入力となる構造特徴を計算し、速度向上と適用性を評価する。

実験結果
リサーチクエスチョン
- RQ1大規模で多様な第一原理の非晶質材料データベースは、組成と温度を横断してLi+拡散度の正確なML予測を可能にするか?
- RQ2非晶系におけるLi拡散度と活性エネルギーと最も強く相関する組成的および構造的特徴は何か?
- RQ3非晶データからLi拡散度を予測する際、アンサンブルモデル(RF、XGBoost)とSISSO記述子はどのように比較されるか?
- RQ4普遍的なMLポテンシャル(M3GNet/CHGNet)は、信頼性のある非晶構造生成および拡散度予測の有用な入力を提供できますか?
主な発見
- データベースは5120組成と79元素をカバーし、5000 Kの非晶集合にはLi含有化合物が3,533件含まれる。
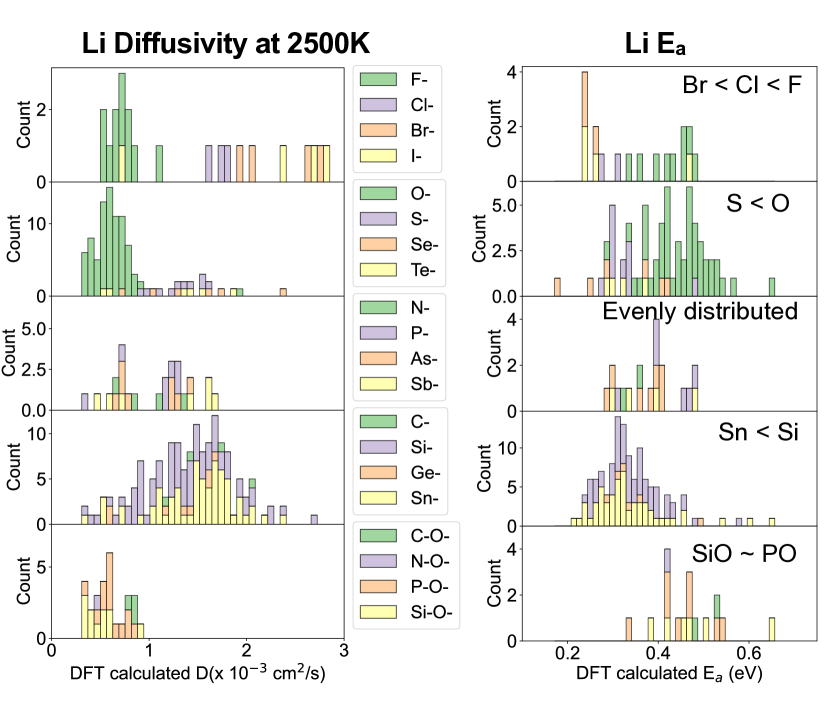
- Li拡散度と活性エネルギーの分布は、アニオン種、カチオンサイズ、電気陰性度差に明確な傾向を示す。
- RFとXGBoostは拡散度予測に対してほぼ完璧な適合度を達成(R^2は1に近く、MAEとRMSEが低く、交差検証で検証)。
- 最上位の予測特徴にはLi%および非Li環境と結合に関する特徴が含まれ、平均結合エネルギー、充填割合、Liの隣接数など。
- SISSOモデルは最適な3記述子で拡散度を予測可能であり、温度・組成・構造間の解釈可能な関係を強調する。
- M3GNetはAIMD由来の構造を高忠実度で再現でき(R^2はRDFで最大0.99)、構造生成を約2000倍加速するが、高温(5000 K)拡散度にはあまり正確でない。
より良い研究を、今すぐ始めましょう
論文設計から論文執筆まで、研究時間を劇的に削減しましょう。
クレジットカード登録不要
このレビューはAIが作成し、人間の編集者が確認しました。